Unlocking the mysteries of the SARS-CoV-2 sugary-coated glycan shield
Toggle
High performance computing has unveiled the atomic structure of the coronavirus’s spike protein, and crucially, its strengths and weaknesses, writes Dr Elisa Fadda, Department of Chemistry and Hamilton Institute
The ongoing COVID-19 pandemic counts over 7.8 million cases and over 430,000 deaths worldwide, according to the World Health Organization (WHO)#1. COVID-19 is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a type of zoonotic enveloped virus, closely related to other forms that have previously caused concern worldwide.
The primary route of infection from coronaviruses involves the interaction between a protein called spike (or S protein) with a protein receptor, called ACE2, expressed on the surface of host cells present in our respiratory airways. The SARS-CoV-2 S proteins stem from the virus’ surface and have been quite realistically represented in many popular graphic illustrations like the horns on a naval mine that trigger detonation.
The name ‘coronavirus’ actually comes from these S protein triggers, resembling the ridges of a crown, or ‘corona’ in Latin. Central to the SARS-CoV-2 S protein’s activity is its ability to ‘open’, exposing a critical domain, namely the receptor binding domain (RBD), to ACE2 for binding. The SARS-CoV-2 S opening represents the key in the door to viral entry and so to infection.
Because of its fundamental role, the SARS-CoV-2 S protein is currently one of the main targets of scientific research worldwide, where the characterization of its architecture and mechanism of action is crucial for the development of specific COVID-19 therapies, such as vaccines, antibodies and antiviral drugs.
To this end, my team in the Department of Chemistry and Hamilton Institute at Maynooth University (MU) and Prof Rommie Amaro’s laboratory at the University of California San Diego (UCSD) joined forces to contribute to the world scientific community effort, by advancing our knowledge on the S protein structure and activity at the atomistic level of detail, through high-performance computing (HPC).
This research approach is based on the use of large supercomputers ranging thousands to millions of cores that can be run in parallel to predict, or simulate, molecular events in real time.
Put simply, molecular simulations can be described as a type of computational microscopy, providing insight into the biomolecular world otherwise unattainable by any other means of investigation. In this specific context, molecular dynamics (MD) simulations provide not only a detailed atomistic description of the S protein’s architecture, but also of its dynamics, stability and mechanism of activation, thus unveiling its strengths and weaknesses.
The complementary expertise of our groups in complex carbohydrates (or glycans) and in large and mesoscale biomolecular simulations resulted in a very large, comprehensive study of the structure and dynamics of the SARS-CoV-2 S protein, Shielding and Beyond: The Roles of Glycans in SARS-CoV-2 Spike Protein.
These simulations involved a very large investment of computational resources, with single experiments counting over 250 computing nodes. The computers were allocated through special COVID-19 initiatives, from the NSF-funded Frontera supercomputer at the Texas Advanced Computing Centre (TACC), now ranked #5 fastest in the world, and the Science Foundation Ireland-funded Kay supercomputer at the Irish Centre for High-End Computing (ICHEC).
Figure 1 below features a 1.7 million atoms SARS-CoV-2 S protein system, observed for the first time through multiple microseconds.
One key feature of the SARS-CoV-2 S architecture that was specifically and uniquely addressed in this study is the role of glycosylation. S proteins are actually glycoproteins, densely covered in a thick layer of complex carbohydrates, also known as glycans, which are too dynamic to be detected by X-ray diffraction and cryo-EM studies, thus generally invisible.
In many viruses, such as SARS/MERS coronaviruses and HIV-1 for example, these highly flexible and branched glycans shroud the surface proteins from the immune system and like a shield, allow the virus to reach the host cell undetected [Watanabe et al, Nat Comm (2020)].
The large scale simulations in our work provide for the first time specific and detailed insight into how the peculiar structure of the SARS-CoV-2 S glycan shield does not only cloak effectively the S protein in its inactive, or ‘closed’, form, but also how it rearranges itself to support and reinforce its active ‘open’ structure, following a ‘lock and load’ mechanism of action.
These results indicate for the first time key vulnerabilities of the SARS-CoV-2 S protein that can be targeted specifically for the rapid development of highly effective therapeutic strategies.
This study of the structure and dynamics of the SARS-CoV-2 S protein, Shielding and Beyond: The Roles of Glycans in SARS-CoV-2 Spike Protein, is available as an OA preprint on the bioRxiv server [Casalino et al, bioRxiv (2020), https://doi.org/10.1101/2020.06.11.146522.
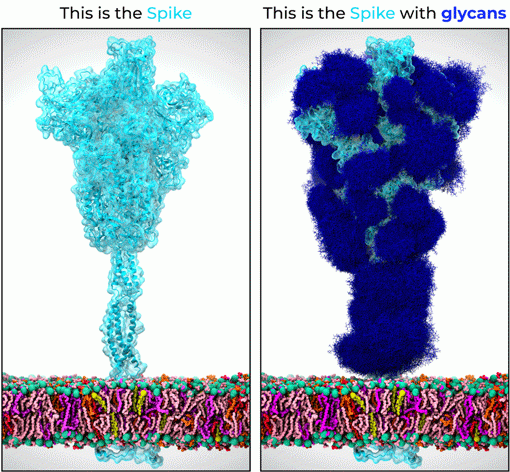
Figure 1. 1.7 million atom 3D model of the SARS-CoV-2 S protein (light blue) embedded in the viral surface membrane (multicolored layer). The panel on the left shows the protein only 3D model of the SARS-CoV-2 S derived directly from experimental data, while the panel on the right shows the correct structure of the SARS-CoV-2 S protein shrouded in its glycan shield (dark blue). Images courtesy of Dr L. Casalino (UCSD). For more information, see Casalino et al, bioRxiv (2020), doi: https://doi.org/10.1101/2020.06.11.146522